






Болезнь Шарко Мари-Тута (БШМТ) – это наследственная патология, характеризующаяся моторно-сенсорной нейропатией (НМСН) хронического течения: атония мышечной мускулатуры, нарушения чувствительности конечностей, а также их деформация.
Заболевание названо в честь медиков, которые впервые описали его в своих научных работах 19-го века: Жан-Мартен Шарко, Пьер Мари и Говард Тут.
Медицинский код заболевания– МКБ-10 – G60.0, включающий в себя группу основных форм болезни ШМТ.
В этой статье мы подробнее рассмотрим терминологию, классификацию заболевания, а также его симптоматику и современные методы лечения.
Терминология заболевания
Как мы упомянули выше, термин «ШМТ» сохраняет в себе фамилии выдающихся враче, которые впервые описали наследственное нейропатическое расстройство периферии.
При этом заболевании человек страдает от полной или частичной атрофии мышц нижних конечностей (дистальных отделов), изменения (деформации) стоп и кистей, сниженной рефлексии сухожилья и нарушенной чувствительностью.
В то же время, основой клинического проявления заболевания ШМТ является поражение периферических нервных волокон, как чувствительных, так и двигательных (моторных), ввиду наследственности. Первые признаки заболевания могут проявиться в пубертатный период или до 25 лет включительно.
Кроме того, «нейропатия» преимущественно обездвиживает мышцы голени, из-за чего, пациент с трудом передвигается и ограничен в движении. Нередко заболевание ШМТ приводит к инвалидизации даже на ранних стадиях обнаружения.

Однако, чтобы как можно быстрее помочь пациенту, приоритетной задачей медиков становиться – определить форму заболевания.
Для этого потребуется детально изучить анамнез пациента, включая период внутриутробного развития, а также генетические предрасположенности.
Помимо подробного сбора информации, невропатолог может провести исследование на проводимость нейронных импульсов в нижних конечностях пациента и развернутый анализ крови с генетическим описанием.
Метод дифференциальной, или всесторонней, диагностики поможет определить форму заболевания и его этиологию.
Врачи отмечают, что заболевание Шарко-Мари-Тута (ШМТ) является наследственным неврологическим расстройством, которое в основном затрагивает периферическую нервную систему. Основные причины его возникновения связаны с генетическими мутациями, чаще всего в генах, отвечающих за миелинизацию нервных волокон. Симптоматика заболевания включает прогрессирующую слабость мышц, атрофию, а также нарушение чувствительности, что может значительно ухудшать качество жизни пациентов. Врачи подчеркивают, что ранняя диагностика и комплексный подход к лечению, включая физиотерапию и поддерживающую терапию, могут замедлить прогрессирование болезни и улучшить функциональные возможности пациентов. Несмотря на отсутствие радикального лечения, современные методы реабилитации помогают многим людям с ШМТ адаптироваться к изменениям и поддерживать активный образ жизни.

Классификация заболевания: Основные формы

- Первая – ШМТ1.
Частота: Наиболее распространённая форма ШМТ.
Наследование: Аутосомно-доминантное.
Проявление: Демиелинизирующая полинейропатия.
Клиническое начало (по факту обнаружения): В возрасте от 10-ти до 20-ти лет.
Клиническая картина (биопсия нерва): Сегментарная демиелинизация (периодически ремиелинизация); форма луковицы; вторичная потеря аксонов. - Вторая – ШМТ 1А.
Частота: До 60% заболеваний группы ШМТ.
Наследование: Аутосомно-доминантное (дубликат периферического белкового соединения – миелина 22).
Проявление: Демиелинизирующая полинейропатия.
Клиническое начало: В возрасте о 10-ти лет от рождения.
Клиническая картина (общие признаки): деформация стоп, атрофия мышц нижних конечностей (голени) и как следствие, нарушение походки, искривление позвоночника. - Третья – ШМТ 1В.
Частота: До 30% заболеваний группы ШМТ.
Наследование: Аутосомно-доминантное (мутация гена MPZ) .
Проявление: Демиелинизирующая полинейропатия.
Клиническое начало: В возрасте до 10-ти лет.
Клиническое проявление подобно ШМТ 1А, однако проявляется более тяжелым течением с полным нарушением опорно-двигательной системы, ввиду деформации конечностей и нарушением проводимости нейронных импульсов. - Четвертая – ШМТ Х1
Частота: Более 10% от всех случаев заболеваемости ШМТ (занимает второе место по распространенности данного генотипа).
Наследование: Аутосомно-сцепленным (Х) (мутация гена, кодирующего конннексин 32).
Проявление: Демиелинизирующая полинейропатия (симптоматика подолгу остается в скрытой форме у женщин, в отличие от мужчин).
Клиническое начало: В возрасте до 25-ти лет (течение патологии у мужчин проходит тяжелее, чем у женщин).
Клиническое проявление сходно с ШМТ 1А. - Пятая – ШМТ 1С.
Частота: До 5% случаев заболеваемости.
Наследование: Аутосомно-рецессивный.
Проявление: Демиелинизурующая полинейропатия с электрофизиологическими признаками.
Клиническое начало: В возрасте от 10-ти до 20-ти лет.
Клиническое проявление: Сходно с Х1, однако проявляются периодические дисперсии проводимости конечностей (в области N.Tibiаlis). - Шестая – ШМТ 1Д.
Частота: Редкая форма ШМТ (до 3% ).
Наследование: Аутосомно-доминантный.
Проявление: Демиелинизующая полинейропатия тяжелого течения (осложнения на всю опорно-двигательную систему).
Клиническое начало: В возрасте от 20-ти лет (позднее проявление симптоматики).
Клиническое проявление: Сходно с ШМТ1, однако течение наиболее тяжелое, характеризуется сколиозом (искривлением позвоночника) с дальнейшим нарушением двигательных функций больного (инвалидизация).
Основные признаки ШМТ: Симптоматика

Зачастую первые признаки болезни Шарко-Мари-Тута обнаруживаются в возрасте от пяти до пятнадцати лет, но, как мы описывали выше, есть формы патологий при которых симптоматика проявляется в более позднем возрасте.
Главные признаки:
- Слабость мышц нижних конечностей, в особенности голени (миопатия, атрофия, атония), ввиду чего, происходит искривление опорно-двигательного аппарата.
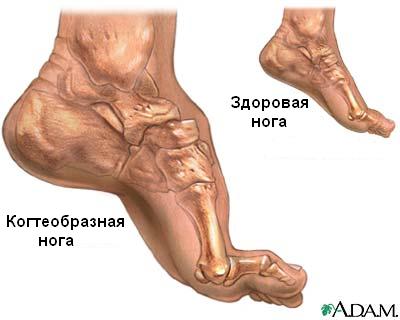
- Структурные изменения (деформация ног, а не только стоп). Может наблюдаться следующая клиническая картина: полая стопа – короткое ахиллово сухожилие, молоткообразное преобразование пальцев ноги, глубокий дугообразный изгиб (смотрите фото ниже).
- Хроническое течение заболевания характеризуется: спотыканиями (из-за деформации стопы, не равномерного распределения массы тела на конечности во время ходьбы), утрудненное передвижение (независимо от скорости: бег или медленная ходьба), невозможность удержать равновесие, стоя на двух ногах.
На заметку. В некоторых случаях миопатия может проявиться в верхних отделах конечностей, например, на уровне бедер и иногда эти проявления путают с признаками простатита.
Кроме того, из-за атонии мышечных тканей может нарушиться моторика рук (невозможность писать, удерживать предмет), а ввиду сниженной проводимости нейронных импульсов, человек теряет чувствительность пальцев к теплу и формам.

Как известно, заболевание ШМТ передается исключительно генетическим путем (от родителей к детям), однако, форма и течение заболевания может отличаться.
Заболевание Шарко-Мари-Тута, или наследственная моторно-сенсорная невропатия, привлекает внимание как врачей, так и пациентов. Люди, столкнувшиеся с этой патологией, отмечают, что основными причинами являются генетические мутации, передающиеся по наследству. Симптомы, такие как слабость в конечностях, потеря чувствительности и деформация стоп, могут проявляться в разном возрасте, что затрудняет диагностику. Многие пациенты делятся опытом о том, как важно раннее обращение к специалистам для назначения адекватного лечения. На сегодняшний день не существует универсального лекарства, однако реабилитационные мероприятия, физиотерапия и поддерживающая терапия помогают улучшить качество жизни. Важно, чтобы люди знали о заболевании и его особенностях, что способствует более глубокому пониманию и поддержке среди близких.

Форма наследования ШМТ на генетическом уровне

Раннее, мы описали основные и самые распространенные виды ШМТ. Как видно, практически все они имеют аутосомно-доминантное наследование. Что это означает? Даже если один родитель 100% здоров, а второй болен, вероятность рождения малыша с ШМТ равна 50%. В то же время, есть два вида патологии, которые наследуются Х-связанным и рецессивным путем, при которых риски значительно ниже. Чтобы определить точную форму заболевания, как у взрослого человека, так и у новорожденного, медики проводят специальный генетических тест.
Диагностика ШМТ пошагово

Диагностика болезни Шарко Мари-Тута подразумевает пошаговую методику исследования.
- Детальный сбор анамнеза пациента и его близких родственников (родители и родители второго поколения).
- Клинический осмотр на силу мышечных тканей, а также чувствительность конечностей.
- Клинический тест (электрофизиологический) на проводимость нейронных импульсов (нервных волокон в конечностях).
- Клиническая оценка неврологического состояния пациента.
Чаще всего врачи проводят анализ ДНК (дезоксирибонуклеиновой кислоты) для установления точной формы ШМТ. Кроме того, при сборе необходимого анамнеза пациент проходит комплексное обследование у следующих специалистов: невролог, генетик, реабилитолог, хирург-ортопед, протезист и другие (при необходимости). Затем, учитывая заключение каждого врача, составляется индивидуальный план дальнейшего лечения (реабилитации или операции, в зависимости от ситуации).
Нередко разные врачи по-своему оценивают состояние пациента, из-за чего их мнения относительно лечения могут отличаться. По этой причине, больным рекомендовано действовать по принципу «третье мнение» и консультироваться с несколькими специалистами одновременно.

Способы лечения ШМТ

На данный момент не существует лечение первопричины заболевания, поскольку его наследование абсолютно генетическое (мутация белков ДНК). Поэтому, наиболее эффективный метод – симптоматическая терапия, при которой задачей медиков является улучшение качества жизни больного и снижением болезненных ощущений во время движения (если оно сохранено).

В первую очередь, пациентам с ШМТ рекомендована санаторно-курортная терапия, лечебная физкультура (ЛФК, направленная на разработку атонических мышц нижних или верхних конечностей при миопатии), применение протезов или отопедических стелек для стоп, корсетов или фиксаторов для голени и ношение специально пошитой ортопедической обуви (под индивидуальных заказ).
Кроме того, в более тяжелых случаях ортопеды-хирурги проводят операцию по исправлению деформации стопы (коррекция свода) или сколиоза.
Перспективы для пациента на лечении

Ученые неоднократно проводили эксперименты, а также длительные мониторинги течения ШМТ у больных животных. На данный момент известно о положительном эффекте современных методов лечения миелинизации нервов периферии у грызунов (ШМТ 1А).
В данном случае, наиболее хороший отклик организма вызвал витамин С, онапристон и нейротрофин-три (антагонисты прогестерона).
Однако, как эти вещества, так и другие все еще проходят лабораторные испытания и не введены в лечение людей, потому говорить о перспективах больных, принимающих медикаментозную терапию – рано.
Также стоит помнить, что любые медикаменты, собственно как и лекарства от простатита назначаются исключительно индивидуально.
Нейротоксины убивают больных ШМТ: Правда или легенда?

Нейротоксины или нейротоксические микрокомпоненты на самом деле есть, и это вовсе не выдумка из голливудского кино.
 Читайте также:
Читайте также:

К ним можно отнести: аурум, нитрофурантоин, пергексилин, таксол, винсистин, метронидазол, адриамицин, метил, этанол, глютетимид, изониазид, дапсон, цисплатин, а также передозировка витамина Д, А, В6 и другие.
Вероятнее большинство из этих веществ знакомы каждому взрослому человеку, поскольку все они регулярно встречаются в качестве компонентов некоторых лекарств и даже продуктов питания.
Нейротоксины вредны для всех людей, и пациенты с ШМТ не исключение – их симптомы значительно усилятся.
Потому, прежде чем начать принимать какие-либо лекарства обязательно следует проконсультироваться со специалистом и никогда не нарушать его предписания относительно дозировки и длительности лечения.
В особенности, мы рекомендуем быть крайне бдительными со следующими препаратами: «Мизомидазол», «Золофт», «Литий».
Подведем итоги
Как мы узнали, болезнь Шарко Мари-Тута – это наследственная патология, характеризующая мышечной атрофией нижних конечностей.
Ее название хранит в себе имена трех французских ученых, которые впервые описали заболевание в конце 19-го века. Существует шесть основных форм ШМТ, которые могут обнаруживаться, как в юном возрасте, так и после 25-ти лет.
Наследование заболевания преимущественно аутосомно-доминантное, однако может быть Х-сцепляемым и рецессивным. На данный момент врачи продолжают работать над созданием медикаментов для качественного симптоматического лечения ШМТ.
И наконец, перспективы пациентов благоприятные, если вовремя начать правильно подобранное лечение (ЛФК, протезы, операция).
Напоследок, рекомендуем просмотреть видеоролик под названием «Болезнь Шарко-Мари».
Вопрос-ответ
Как передается Шарко Мари Тута?
Пути наследования: аутосомно-доминантный (чаще всего), аутосомно-рецессивный, Х-сцепленный.
Как вылечить болезнь Шарко Мари Тута?
Лечение не может остановить прогрессирование болезни Шарко-Мари-Тута. Для компенсации симптома свисающей стопы можно использовать шины, иногда для стабилизации стопы показана ортопедическая операция. Для борьбы со слабостью полезна физиотерапия и занятия по поддержанию двигательных навыков.
Какие симптомы характерны для невральной амиотрофии Шарко-Мари?
В отдельных случаях невральная амиотрофия манифестирует расстройствами чувствительности в стопах, наиболее часто — парестезиями в виде ползания мурашек. Типичным ранним признаком ШМТ является отсутствие ахилловых, а позже и коленных сухожильных рефлексов.
Как диагностируется зуб Шарко-Мари?
Биопсия нерва . Небольшой кусочек периферического нерва извлекается из голени через разрез на коже. Лабораторный анализ нерва отличает болезнь Шарко-Мари-Тута от других нервных расстройств.
Советы
СОВЕТ №1
Обратите внимание на ранние симптомы. Если вы заметили слабость в мышцах ног, затруднения при ходьбе или изменения в чувствительности, не откладывайте визит к врачу. Раннее выявление заболевания может значительно улучшить качество жизни и замедлить прогрессирование болезни.
СОВЕТ №2
Изучите генетическую предрасположенность. Заболевание Шарко Мари-Тута может быть наследственным, поэтому важно узнать о семейной истории. Если у вас есть родственники с подобными симптомами, проконсультируйтесь с генетиком для оценки риска.
СОВЕТ №3
Поддерживайте активность и физическую реабилитацию. Регулярные физические упражнения, адаптированные к вашим возможностям, могут помочь сохранить мышечную силу и улучшить координацию. Работайте с физиотерапевтом для разработки индивидуальной программы реабилитации.
СОВЕТ №4
Обратитесь за поддержкой. Заболевание может оказывать значительное влияние на эмоциональное состояние. Не стесняйтесь обращаться за помощью к психологам или группам поддержки, где вы сможете поделиться опытом и получить советы от людей, столкнувшихся с аналогичными проблемами.



